
Jeffrey Moffitt, Alistair Boettiger, Xiaowei Zhuang
jeffmoffitt@gmail.com
alistairboettiger@gmail.com
Version 2.0: October 2013
Purpose and Overview
This protocol describes the construction of OligoPaints FISH probes from a complex oligo library via reverse transcription of an RNA intermediate. By exploiting the high yield of in vitro transcription, the high efficiency of reverse transcription, and the selection hydrolysis of RNA in alkaline conditions, this protocol creates ~100 pmoles of single stranded DNA probe without exceeding a reaction volume of 50 uL. The protocol can be completed in 24 hours, and requires only a few hours of hands on time.
Protocol
1. Template Construction
Purpose: Construct DNA templates for the in vitro reaction.
- For each sub-library, mix the following in a single PCR tube or well of a PCR strip or plate
- 2.5 uL 10 uM Universal Primer
- 2.5 uL 10 uM Index Primer with T7 site
- 1 uL 100 pg/uL complex oligo library (adjust amount as needed)
- 19 uL ddH2O
- Mix well and add 25 uL 2xPhusion Master Mix
- Run the following thermocycler program
- 98 C; 3 minutes
- 98 C; 5 s
- 72 C; 15 s
- Go to 2; 29 times
- 72 C; 2 minutes
- 4 C hold
- Clean up each reaction according to the instructions in the Zymo DNA clean and concentrator manual and elute each reaction with 15 uL ddH2O
- Optional: Step 4 can be done for multiple reactions in parallel by using the plate purification kit
Notes: We prefer Phusion polymerase over less expensive polymerases because it works robustly in our hands and also because we can anneal our primers at higher temperatures (72C as part of the two-step annealing process).
The T7 site should be added to primer that corresponds to the anti-sense strand to the final probe strand. We use the following T7 promoter sequence: TAATACGACTCACTATAGGG. Thus, a typical reaction might involve the universal primer, P9, and the T7 index primer, P2_T7.
P2_T7
TAATACGACTCACTATAGGG GCCCGCTCTTGTATCGGACC
P9
CAGGCATCCGAGAGGTCTGG
2. Template Quality Control: Optional
Purpose: Confirm that the template PCR worked before proceeding with the protocol.
- Mix 1 uL of template with 9 uL of 1x Sodium Borax
- Add 2 uL of 6xOrange Loading Buffer with 12x GelGreen
- Load on a 2% w/v agarose gel made with 1x Sodium Borax
- Create a ladder sample using the same proportions of buffer and ladder as above
- Run the gel at 300V for 15 minutes in 1X Sodium Borax
- Image
- Nanodrop the sample to determine the yield
- Typical yields vary between ~0.5 – 2 ug
Example:
- Typical yields vary between ~0.5 – 2 ug

Figure 1: Image of a 2% agarose/1X sodium borax gel with the templates for six different sub-libraries. The ladder is the GeneRuler Ultra Low Range ladder.
The shortest band is the expected length for this oligo library (84 nt). The next largest band, which is the darkest, corresponds to what we term a ‘bubble’ product. In the final stages of the PCR, when the PCR mix is exhausted, molecules in the library tend to reanneal to the incorrect molecule. These products contain properly basepaired sequences at the common 5’ and 3’ priming sequences but are not properly base paired in the probe region. This molecular bubble produces the slower migration on the agarose gel. Since T7 needs only a double-stranded region in its promoter, these molecules are still suitable templates.
3. Construction of Reverse Transcription Template via In Vitro Transcription
Purpose: Construct RNA templates for the reverse transcription reaction.
- For each sub-library, mix the following in a single PCR tube or well of a PCR strip or plate
- 10 uL DNA template
- 2 uL 10X T7 Reaction Buffer
- 2 uL 100 mM ATP
- 2 uL 100 mM CTP
- 2 uL 100 mM GTP
- 2 uL 100 mM UTP
- 1 uL Murine RNase Inhibitor
- Mix well via pipetting and add
- 1 uL T7 RNA polymerase mix
- Mix well via pipetting
- Incubate at 37C on a thermocycler for 16 hours
- Optional:
* Clean up each reaction according to the manual for the Zymo RNA clean and concentrator-100 kit. Elute in 100 uL DEPC-treated ddH2O
* OR Purify using RNA clean XP-beads from Beckman following manufacturer’s protocol for tubes or plates.
* OR Purify the reactions using the plate version of the RNA clean and concentrator (small yield only!)
7. Optional: Nanodrop the reaction to determine the concentration and yield
* Typical yields vary between 40 and 100 ug
4. Construction of Probes via Reverse Transcription
Purpose: Create the final ssDNA probes.
- For each sub-library, mix the following in a single PCR tube or well of a PCR strip or plate
- 10-20 ug RNA template
- 4 uL 100 uM labeled primer
- 8 uL 10 mM dNTP mix
- DEPC-treated ddH2O to 26 uL
- Incubate at 65C for 5 minutes, then mix in the following
- 8 uL 5X First-Strand Buffer
- 4 uL 0.1 M DTT
- 1 uL Murine RNase Inhibitor
- 1 uL SuperScript III
- Mix via pipetting and incubate at 42C for 50 minutes
- Heat inactivate at 72C for 15 minutes
- Add the following to each reaction
* 10 uL 0.5 M EDTA
* 10 uL 0.5 M NaOH
7. Incubate at 95C for 10 minutes to destroy the RNA template
8. Clean up the reaction using the Oligo Clean and Concentrator kit according to the instructions in the manual. Elute in 20 uL ddH2O.
Notes: The primer for the reverse transcription can be used to add fluorophores or secondary sites. In the example reaction discussed above, we would use a Cy5-labeled universal primer.
P9-Cy5
/5Cy5/CAGGCATCCGAGAGGTCTGG
5. Quantification and Quality Control of Probes
Purpose: To determine the fraction of the primer incorporated into the probe and, thus, the probe yield and to determine if the reaction was successful.
PAGE: Check probe integrity and length
- For each sub-library, mix the following in a single PCR tube
2 uL probe
8 uL ddH2O
10 uL 2xTBE-Urea Loading Buffer - Prepare an identical sample using the GeneRuler UltraLowRange Ladder
- Heat samples at 95C for 5 minutes then incubate on ice
- Prepare a 15% TBE-Urea Precast Gel and run at 120V for 1 hour
- Optional: The gel box can be placed in a 60C water bath to maintain a uniform temperature and improve the quality of the gel
- Remove urea from wells and add 20 uL of each sample
- Run the gel at 120V for 50 minutes
- Remove and stain in 1xTBE solution with 10X GelGreen for 15 minutes
- Wash gel in 1xTBE briefly and image
Nanodrop: Estimate the yield of full length probe
- Make a control/standard:
- Dilute 1 uL 100 uM of the labeled primer into 19 uL ddH2O
- Nanodrop each library member and the standard
- Estimate the expected absorbance for the full length probe from its molecular weight
- Assuming the expected absorbance for full length probe represents 100% incorporation and the measured standard represents 0% incorporation, use linear interpolation to estimate the fraction of full length probe.
- Estimate the final concentration of probe from this fraction and the total absorbance at 260 for each probe
Example:
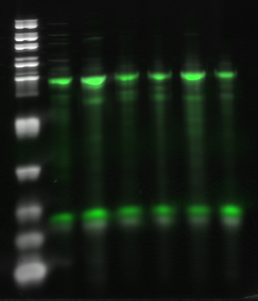
Figure 2: Overlay of a GelGreen image of a 15% TBE-Urea PAGE gel (white) and a Cy5 image of the same gel (green). The six bands represent 6 different sub-libraries in which 100 pmol of labeled primer was used to reverse transcribe 5-10 ug of RNA template in a 20 uL reaction volume. Primer (bottom band) was incorporated into probe (top band) with an efficiency that ranged from 45-95%. Incorporation efficiencies calculated from the intensity of the Cy5 labeled bands and from the absorbance spectra agree well.
Main Components
- Phusion High Fidelity Polymerase Master Mix (NEB M0530S/L)
- T7 High Yield RNA Synthesis Kit (NEB E2040S)
- SuperScript II Reverse Transcriptase (Invitrogen 18064-014)
- Murine RNase Inhibitor (NEB M0314S)
- DNA Clean and Concentrator-5 Kit (Zymo Research D4003 or D4004)
- RNA Clean and Concentrator-100 Kit (Zymo Research R1019)
- Oligo Clean and Concentrator Kit (Zymo Research D4060)
- DEPC-treated Deionized Water (Invitrogen AM9922)
- 0.5 M EDTA
- 1 M NaOH
- 15% Precast TBE-Urea gels (Biorad 456-6055)
- 2X TBE-Urea Loading Buffer (Biorad 161-0768)
- GeneRuler Ultra Low Range Ladder (Thermo Scientific SM1212)
- UltraPure Agarose (Invitrogen 16500100)
- Sodium Borax (Sodium tetraborate; Sigma 221732)
- GelGreen (Fisher NC0422689)
- 6x Orange Loading Buffer (Fisher R0631)
- Eppendorf Tubes, 1.7 mL
- Table top microcentrifuge
- PCR strips, tubes, or plates
- Thermocycler
- Gel electrophoresis equipment for both agarose and PAGE
- Nanodrop
- Complex oligo library (CustomArray)
- Appropriate primers with labels (IDT)
Optional Components: Plate Purification - ZR-96 DNA Clean and Concentrator (Zymo Research D4017)
- ZR-96 RNA Clean and Concentrator (Zymo Research R1080)
- ZR-96 Oligo Clean and Concentrator (Zymo Research D4062)
Accessory Buffers - 10X Sodium Borax
- Dissolve 10 g of sodium tetraborate in 1 L water.
- Lightly boil in a microwave to fully dissolve powder
- This buffer is an alternative electrophoresis buffer that allows gels to be run in a fraction of the time. TAE can be substituted throughout if desired
- To make 1x buffer, simply dilute 9:1 in water.
- 6x Orange Loading Buffer with 12x GelGreen
- Dilute 1.2 uL of 10000X GelGreen into 1 mL 6x Orange Loading Buffer
- This mix allows staining of DNA samples in agarose to be done during the run of the gel with the use of minimal stain