
12:30 pm – 7:00 pm
(sick)
RNAi experiments
Primer issues
- diagnosing problems with primers for qPCR
- writing code for primer mapping
- organizing a better data structure to keep track of all the RNAi targets, their qPCR primer sets, their RNAi primer sets, their accession numbers, etc.
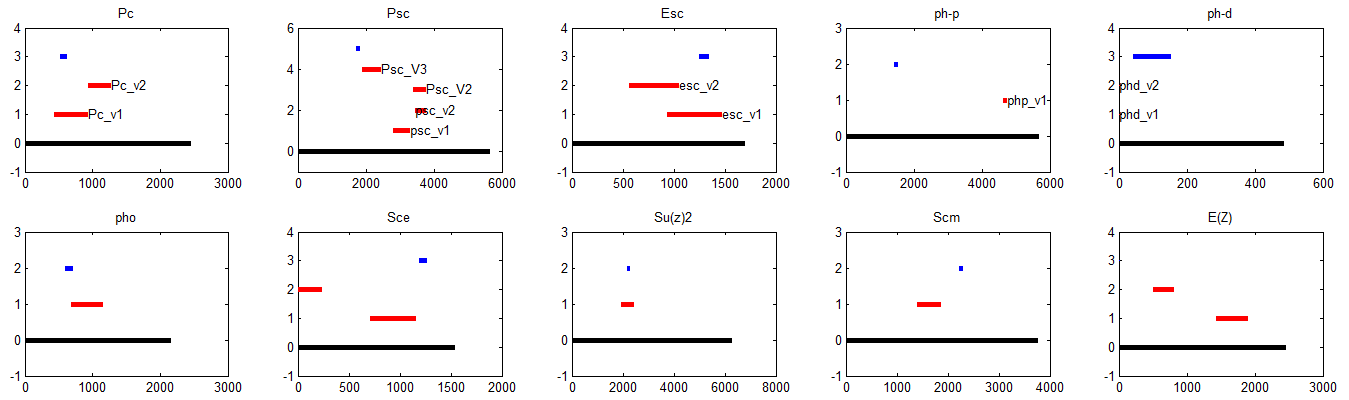
- my overlap checking clearly did not work correctly. Overlap of qPCR and RNAi primers detected for RNA
- additionally, several of the 20mer primers have perfect hits elsewhere in the genome. Only one should have a sufficiently close compliment to be able to amplify so it shouldn’t be a problem, but still, would have been good to check sooner

- note Pc_v1, Esc, Pho, and Su(z)2 qPCR primers all overlap RNAi
- and apparently the polyA selection for cDNA conversion does not sufficiently screen out the added RNA
RNAi new knockdown
- 2 6-well wells with Ph-p + Ph-D (30 mg/mL)
- 4 mocks
- used cells from SFX, rinsed in PBS and repelleted
- one 75mm2 flask, could have used a few more cells for a bit better density, but it is okay.
- incubated 55 min.
Cell culture
- passaged Psc- cells with Trypsin.
- washed cells with PBS to remove excess serum (inhibits Trypsin)
- added 3 mL Trypsin soln (now in 4C).
- After 20 min, they blow off easily. (After 10 they just start rounding up. Next time try just 10 min)
- add a bit of media to try and inhibit trypsin and pellet cells
- wash in 5 mL PBS (resuspend and repellet)
- resuspend and plate in 75mm flask in fresh media.
Embryo in situs
- remade hybe buffer, dissolving BSA in H2O first (never went into solution otherwise).
- hydrating embryos into PBT
- diluted probes (5 nmol) to ~50 uM in ddH20, 50 probes so individual probes at 1 uM. recommended final concentration at 1 nM per probe. Lets try 2 uL in 20 uL hybe buffer O/N. Thats ~10 nM.