
10:00 A — 8:00 P, 9:15 P – 11:30 P
OligoPaint Probes
new PCR
- Pool samples: 4 mL Ubx, AbdA and AbdB, 2 mL each B, Y, K and G.
- cooling down ultra-centrifuge (takes 3 hours, good thing to start while samples are precipitating in freezer)
- added 16 uL glycogen to all samples, + 100 uL 4M NH4oAc per mL to each sample. Vortex
- added 13 mL 100% cold EtOH to each 4 mL scale reaction and 7 mL to each 2 mL scale reaction
- freezing at -80C, ~3P
- Test gel of PCR product. (First gel ran backwards, box direction was flipped). Ran out 3 uL of a 1:10 dilution.

- Y and K don’t look good. Redoing reactions with 2x primer and 2 uL per 100 uL of PCR 1 stock (short primer amplicons at ~100ng/uL)
- spin down in ultra-centrifuge: 14,000 xg, -3C, time = hold (~ 1 hour)
- wash with 70% ethanol, respin.
- Spin in swinging buckets moves pellet to bottom of the 50 mL flask, where it can be more easily resuspended. Also helps pellet stick to facilitate removal of ethanol
- Low glycogen precipitations definetely have more fall-apart pellet (using 1/5th recommended amount in all samples).
- let drip dry upside down ~ 3 hrs. Dries well.
- Resuspend 4 mL scale in 488 uL of ddH2O and 2 mL scale reaction2 in 244 uL ddH2O.
- Y and K look good this time (see on T7 gel).
- Set up 5 mL scale of Y and K PCR to run O/N.
- Still need to nicking enzyme to arrive from NEB.
- B, Y and K primers are out. Should re-order sometime at the 50 ng scale.
Troubleshooting
- Incomplete nicking: maybe nick sites are mutated in library? If PCR primers extended in to overlap the nick sites, we can should be able to correct possible mutations
- Ordered Nicking primers including nick sites (will better match the adapter Tm anyway).
T7
- Set up 10 uL scale reactions (1 uL T7, 1 uL NEB buffer 4, 8 uL resuspended DNA)
- Gel
*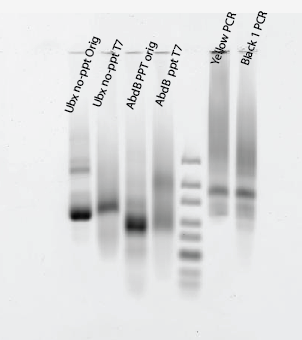
- Maybe worked?
- No-PPT slowly being digested, no change to shape, intensity drops,
- PPT being converted into single strand, runs as smear
- Neither complete. Hard to tell, need better controls:
- Exonuclease digestion, next steps:
- Compare different enzymes
- RecJ{f}
- T7 exonuclease
- negative control PPT modified reverse primer — should be undigestable if both are PPT
- positive control pure doublestranded DNA (no bubble library stuff).
OligoPaint Hybe optimization
- Analyze dot intensity and background signal for 4 different hybe temperatures
- working on summary slides (Hybe_analysis.odp)
- BX-C looks qualitatively different in Clone8 cells
STORM observations
- from yesterday
- My slide tweezers got a slight spot of rust on them, and an almost invisible fleck stuck to coverslip 3, which I did not notice until the slide was sealed in epoxy. But the little bit or iron reacted with and destroyed the buffer, so while the signal looked good, none of the molecules photo-switched at all.
Ph-FLAG
- Next round of experiments
| Cell line | Primaries | Secondaries |
|---|---|---|
| wt Ph-FLAG | m a-flag + rb a-Ph | dk a-m 647, dk a-rb 750 |
| wt Ph-FLAG | m a-flag + rb a-Ph | dk a-m 750, dk a-rb 647 |
| M1 Ph-FLAG | m a-flag + rb a-Ph | dk a-m 647, dk a-rb 750 |
| M1 Ph-FLAG | m a-flag + rb a-Ph | dk a-m 750, dk a-rb 647 |
| S2 cells | rb a-Ph | dk a-rb 647 |