
8:40a – 1:15a
Ph project
- Get S2 cells for Ajaz
- Running more simulations to explore appropriate range and parameters for the effect of PhM concentration on clustering.
- For the moment let’s ignore the unbound populations of molecules. Surprisingly I observe a saturating effect of PhM concentration happening fairly early / at relatively low levels of PhM (e.g. around 300, with wt levels around 500).
Meetings
- Lab meeting (see protected notes)
Project 2
- helping Hao with alignment issues in earlier data set
Chromatin project meeting prep
- Goals for BB for tomorrow: area and other parameters as a function of region size.
- Analyzing E10 data. average error in drift correction generall 2 – 5 nm
Deep sequencing
- number of wrong primers 2000 (C05) 20000 F03 (currently ). This does not distinguish wrong primers from wrong sequences
- writing new sequence to library parser (hopefully faster) using the read command.
- New
MapLibraryReads.m
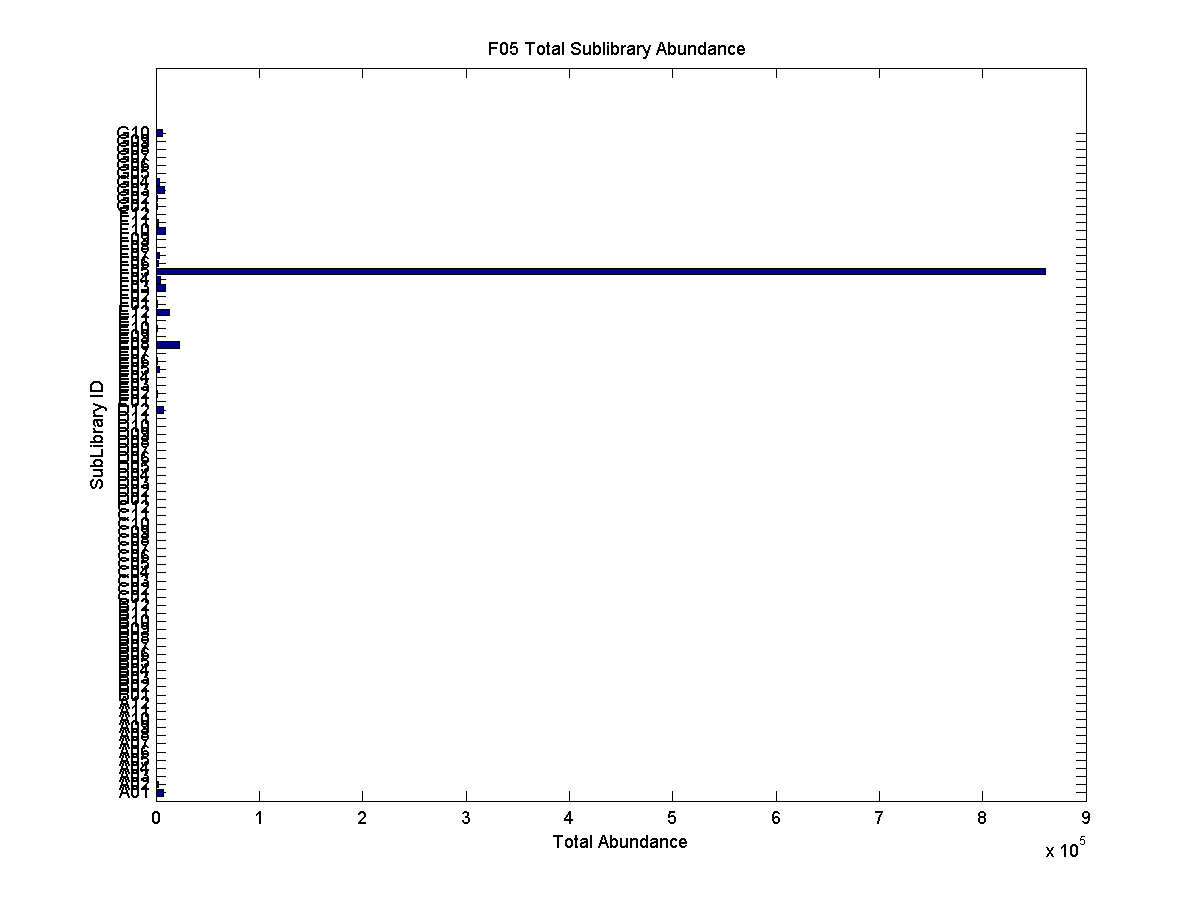

Sample Case: 156 probes (index 3). This sub-library hardly stains cells at all.
* only 3% of total reads with this index map back to this sub-library
* ~80% of the reads that map to the correct library have the expected full length (88 bp)
* the other sequences are highly variable in length (25-63 bp) due to internal, off target priming. (as hypothesized last night). In fact it looks like the unintended cross talk is 100% driven by internal priming, since there are 35 bps between the longest off-target sequence and the expected length. If the orthagonal primer sequences had hybridized I would expect 78 bp matches.
* More probes come from the correct sub-library than from any other library, but only by a fraction. The other sub-libraries at frequencies which directly correlate to their size/complexity (as expected).
So there must be a not-to-obvious smear of smaller stuff in my gel that is propagating, and these will go on to make RNA and ssDNA probe since they have the correct T7 site from the index primer.
But maybe we can exploit the fact off-target contamination is driven by internal priming with a good gel purification could separate the 88 fragments from the <60bp fragments. The column PCR cleanup probably already gets rid of the small fragments (20-40 bp?). (I did not run a column clean-up on the PCR samples I sequenced).
I think this also explains why the small libraries amplify much better using a 20bp index primer without a T7 site than they do with the 40 bp T7 primer. There is more total primer DNA in the mix to make off target interactions?
Which also argues that maybe dropping the primer concentration (a lot?) for an initial PCR may pull out the rare library with more fidelity (it might take a lot longer before we start seeing amplification, but hopefully we get less off target amplification drowning out the signal). Maybe after a bunch of rounds of amplification with extremely low primer concentration, I can bump the primer concentration back up and do more PCR and get a purer product? Or does that just sound ridiculous?
Things look much better for longer probes.