
9:15a -10:00p
Goals
- Restart Fasta file building for genomics
Coding
- removing GPU from STORMrender
- not compatible with CUDA 5.5
- not restorable after CUDA update
- not distributable / not multiplatform
Genomics
- Computer reboot terminated building gene list, around 60% complete
- Relaunched on Tuck, with directions to write genes to fasta file as it goes. This way if it gets aborted we can restart from where it left off.
- may take 2 – 3 more days running on Tuck.
- Too much memory used. Cancelled run on Tuck (1.2% complete).
- Restarted on Monet
- Monet rebooted to install new CUDA
- Relaunched analysis. (CUDA openMM failed).
STORM
- Finish O/N STORM of D08
- Set up O/N STORM on next sample on STORM2: E11 (50kb Green, scattered repeats).
Chromatin region analysis
- Remake G5, test in new modENCODE sequenced Kc cells
- Regions to test sequencing results:
- G4 (failed repeatedly)
- F3, F4, F5 (haven’t made succesfully)
Sequence analysis
- Note: saved index is reverse-complement of the last 20 bp
- chop of indices and keep sequence in between, align that to sub-library.
- Library purity under-estimated: reads with just outside primers all reported as off target reads.
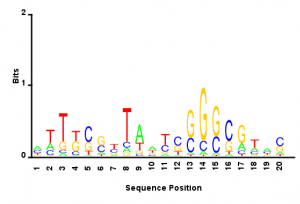
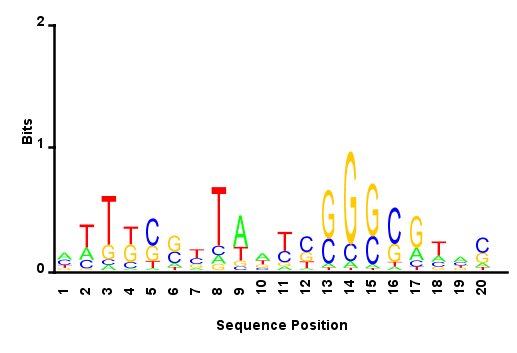
- Reads from E06 sublibrary that have long off target inserts (>30 bp) have the E06 primer cross hybridized most frequently to something that vaguely resembles the T7 primer: note the ‘GGG’/’CCC’ and ‘ATTA’/’TAAT’ regions.
- all told there are a bit less than 17,000 of these long insert off target sequences.
More observations
- cutting to 10 bp or longer inserts the motif is less pronounced. (373,000 sequences)
- So if I restrict my analysis to reads that have 10+ bp of sequence in between the common primer and the index primer as you suggested, I can validate that the short reads largely do result from mispriming on other sublibraries. (This is easily demonstrated by aligning the non-primer sequence of 10+bps back to my library as we discussed). I take this as good news because it library cross-talk should be correctable by dual indexing. (When I found a lot of reads with almost nothing in-between I was concerned that additional problems aside from sublibrary cross-talk plagued the short libraries, which might not be correctable by dual indexing).
- This lets me return to the motif question: Combining all the reads which result from mispriming of the index 10bp or more away from the end of the common site, (10 bp+ of sequence gives me a little more confidence that the reference has been mapped correctly) I get a weak motif (see attached). Doesn’t seem to suggest a strong sequence bias. If I look at the random nucleotide sequence added on in the adapter to help control for PCR amplification bias skewing the sequence the diversity is very high. So what motif like nature there is to this logo is probably not an artifact of the sequencing prep PCRs.

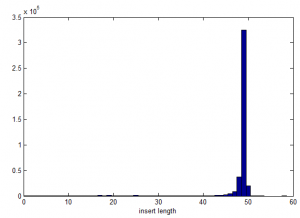
Insert length
- E06 vs F03
- (perplexing side note: F03 doesn’t give good spots).


New primer screening
- oligoprop all index-T7 fusions
- no correlation between difficult to get spots and T7/index dimer hairpins
Cell staining
- Test new short probes (8 hybes)
- On deck: new RNA stains
- On deck: restain G5 (with RNase)