
10:00 am – 6:00 pm,
New Library synthesis
- chr-L8 (en-locus)
- T7 L6E3_R primer_232 TAATACGACTCACTATAGGG TTGCGCGAGACCAACGTACG
- L6E1_F primer_226 CCGTACGTCGAGTCGGGTCG
- Was: T7 L6E2_R primer_230 TAATACGACTCACTATAGGG TGATCATCGCTCGCGGGTTG, swapped out for L6E3_R to avoid cross-talk with DSB-lib
- Also doing L7 10 kb and 2 kb (to make in cy3).
per reaction
- 25 uL enzyme-master
- 2.5 uL 1:40 library
- 2.5 uL F
- 2.5 uL R
- 2.5 uL EvaGreen
- 10 uL ddH2O
Master
- 100 enzyme + 10 Evagreen + 40 ddH2O, 40 ea.
- sample order: tab, L8 (en), L7-10kb (BXC), L7-2kb (BXC)
- out of 10x Sodium Borax (Na-Borax). 10g Sodium Tetraborate in 1 L ddH2O
Column cleanup
- per instructions, on DCC-5
- ran gel. Order: L8a, L7 10 kb, L7 2 kb, L8 try 2
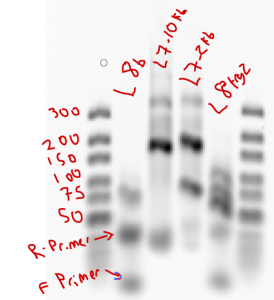
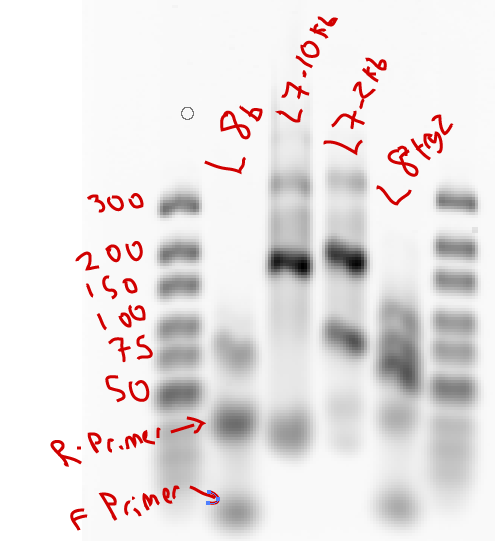
Observations: L8 failed both times (amplified up through cycle 30 first time, didn’t start until cycle 25, started around cycle 22 when run at 62C annealing instead of 65C for second run, still stopped at cycle 30). L8 probes amplified by cycle 25 (later than last time). Bands of L8 look decent (should be 142)
Re-run
- redid L8 using common primer. Amplified by cycle 15-16 (stopped at 22, a bit late).
- proceeded with gel cleanup.
- ran gel with residue from PCR tube after running cleanup (low conc)

Communication notes
- write to Cornell
- update Stanford
- write to XZ – re: new collaboration
Data requests
- sent volume-rg data to Marc M.R.
- sent molecule lists data for docking models to same.
- requested export to excel files, need to do today – processing, export from matlab to excel is slow. sample data for first 3 domains looks good.