10:00 am – 10:00 pm
Multicolor analysis
Chromewarps
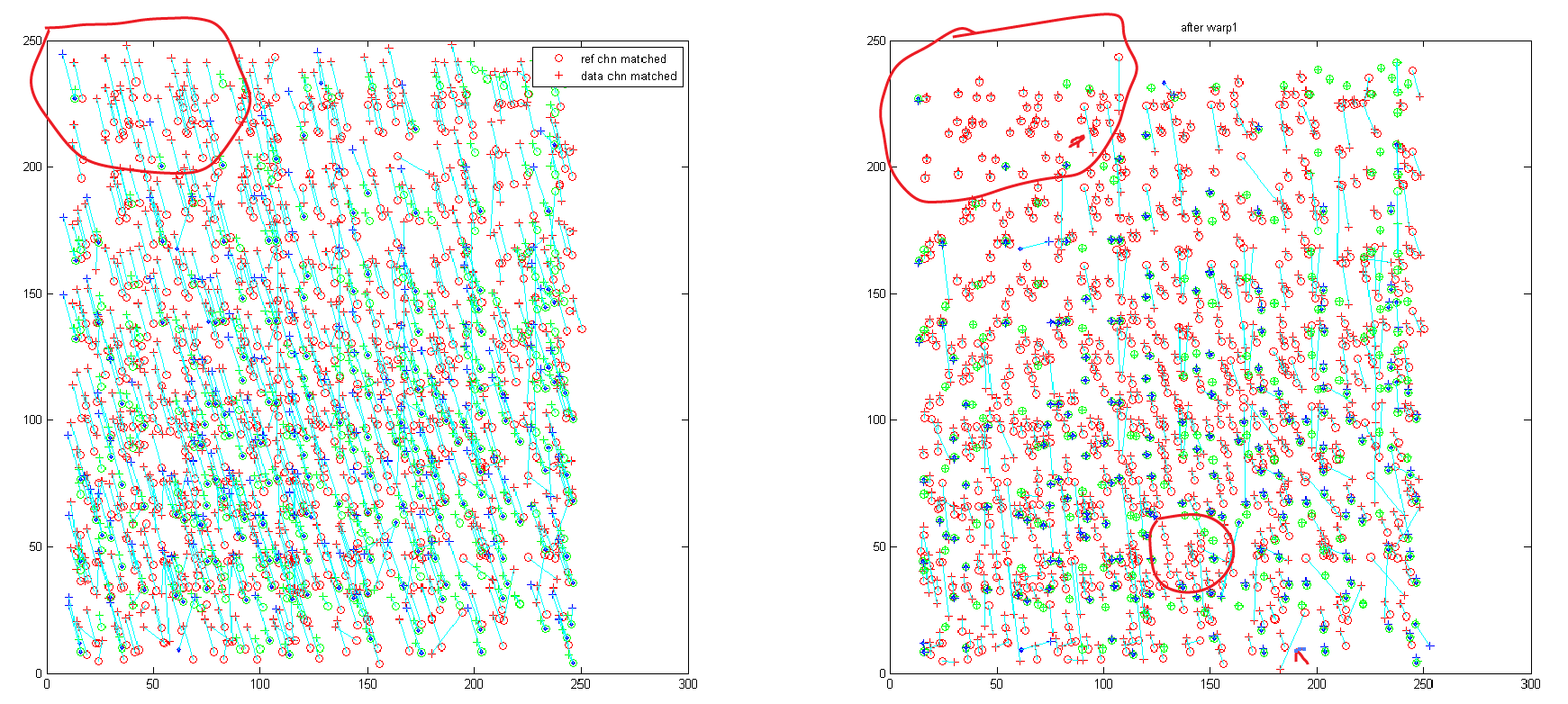
- This code is still not too robust. A few mismatched outliers constrain and skew the quality of the warp map.
- It also is harder when beads from a single field are very dense and when the quadview is badly misaligned.
- CS always realigns the quadview before using the microscope and normally after that it aligned to within a pixel, even if it was off by 10s of pixels before. This time it was already aligned within a pixel and when I got back to the scope it was evidently out of line by 10s of pixels! My code should be able to handle this just fine (with some substantial reduction to the usable field of view, 30 pixel misalignment means 60 of 256 pixels lost)
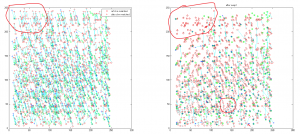
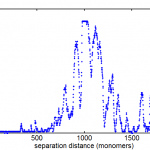
- proposed fix: lets do two warps, and throw out all the outliers before the second warp. Their are very clearly two populations, one that has matched quite well, and one that is off by anything from 10s of nm to 1000s of nm.
- or maybe better, just rematch references and data after the first warp with a more aggressive cut off — I believe this was the original way anyway.
- also, 3D correction via 3D bead fitting is out, so lets remove it from the ChromeWarps code. This is better corrected by adjusting the imaging plane offset between the movies so all the data is already maximally in focus and the zero points of the z-fits are already aligned to match. differences in the zero-point z-positions can be measured with z-calibration files
- currently I determine this by eye to be about an 80 nm offset, which could be off +/- 40nm
- I should probably try this with the same set of beads and fit both movies
- Also should have z-calibration code return the offset from the starting point — not sure I save this as an output right now of if the graphs get re-centered.
- Final solution:
- MatchFeducials autocorrelation approach with upsampled images can run into trouble sometimes (not at all clear why, possibly this moves big shifts outside of the default region size for corrMmini). We totally don’t need sub-pixel registration for this initial step to just get the x,y shift so I changed the default to 1 pixel (no up or down-sampling).
- also had bug in finding unique nearest-neighbor matches between molecule sets, which had been hacked with a final distance cutoff to reject the occassional weird errors. This has now been fixed.
- If using autocorrelation, the cutoff distances should be unnecessary. As long as enough nearest neighbors are unique everything is good, it doesn’t matter how many total neighbors you have inside the search window.
- only parameters.AffineRadius has any function now, parameters.matchRadius is not used.
- updated solution pushed to release (ZhuangLab)
Data analysis
Chromatin simulations
now launch to python. Then add openmmPolymer to path and import polymerutils
import sys
sys.path.append("/n/home05/boettiger/OpenMM/openmmPolymer")
import polymerutils
import knotAnalysis
data = polymerutils.load("/n/home05/boettiger/OpenMM/Data/myPoly0082")
print data
num = knotAnalysis.analyzeKnot(data)
print num
local coding:
- output of
polymerutils.grow_rw cannot be inserted straight into Simulation.data
- hacked this by converting to a list of lists and multiplying that by
unit.nanometer (some weird class of its own from simtk). this object can successfully be passed to Simulation.save and read back in with Simulation.load (after which it counts members and populates Simulation.N). So after loading this object as an initial polymer structure the simulation will run without errors.
- This approach is implemented in
MirnyConfinedStickyTemplateV4.py
- I tested the inital structure and one of the later timesteps from this simulation with 82 monomers. Both return
num = 1 under knotAnalysis.analyzeKnot run on Odyssey (requires linux platform). I was expecting 0 for uknotted but apparently this code does 1.
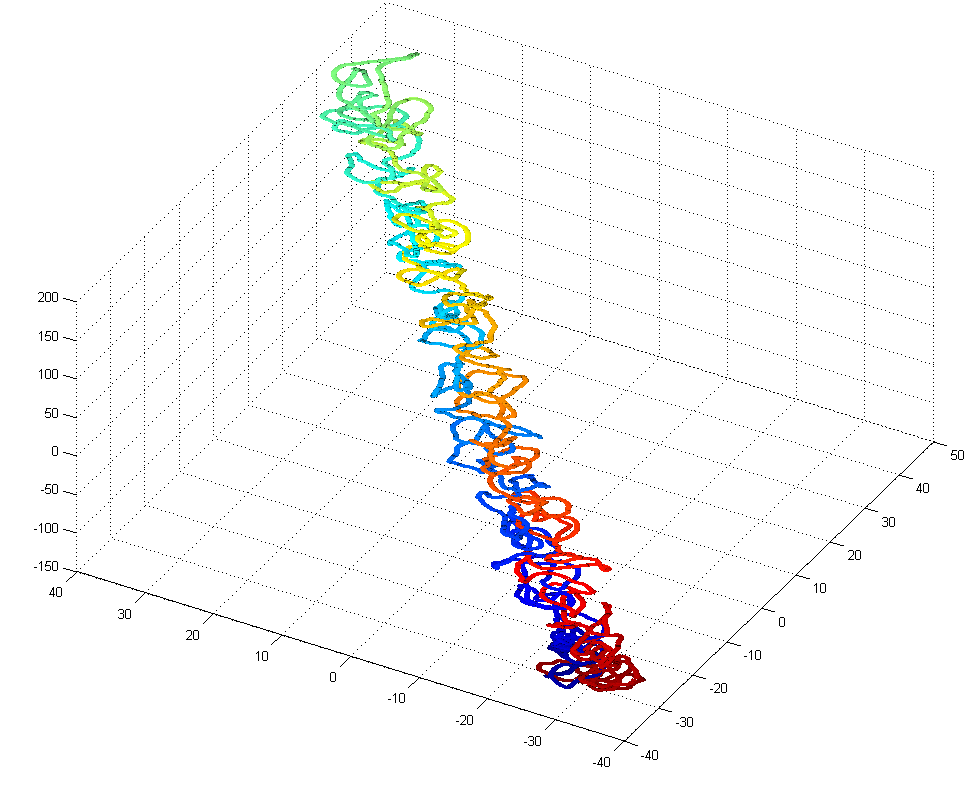
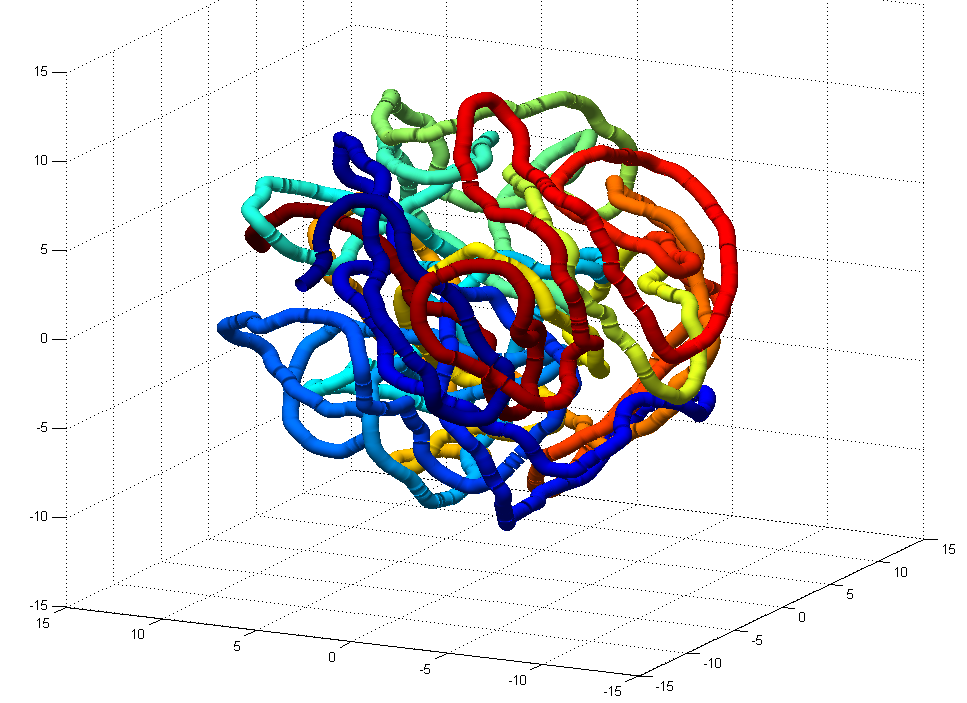
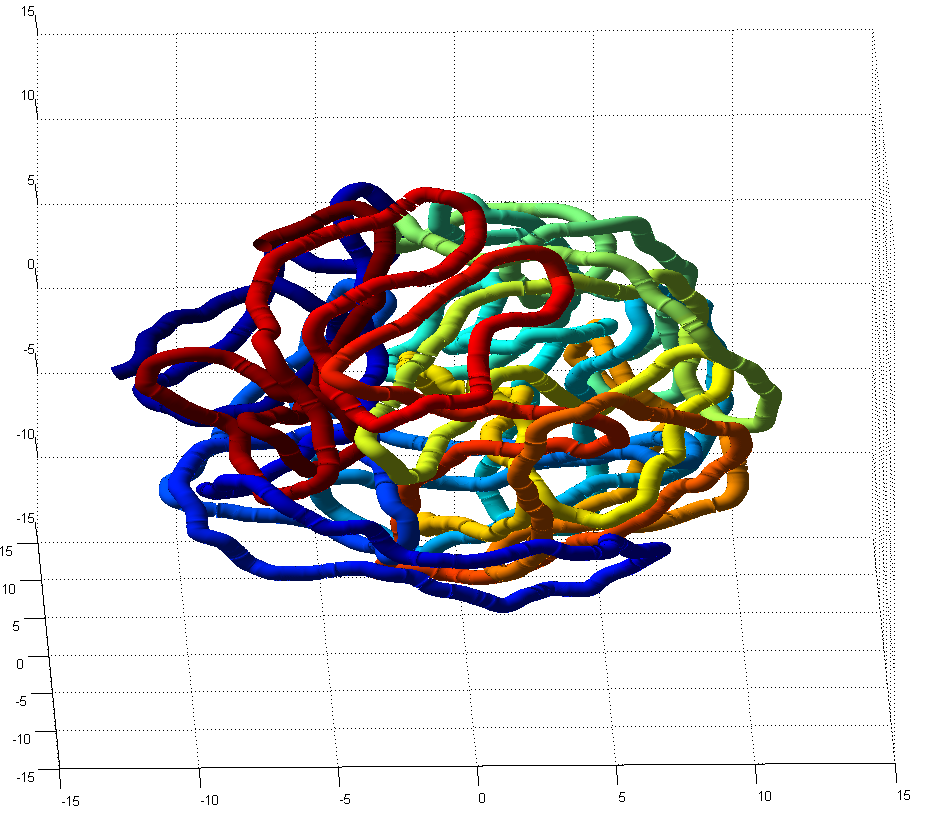
- this could use some further validation. Visual inspection at least makes it look believably unknotted.
- this is at least much better than my previous 200 node random-walk polymer that returned a non-integer value larger than the number of nodes, and had what appeared to be near-certain knots by visual inspection / rotation of the 3D polymer.
More Odyssey coding
- running code failed at import
simtk.unit as unit
- attempted to
pip install simtk in conda. Failed.
- running on gpu nodes: should look something like this
srun -p resonance --pty --mem 500 /bin/bash
- I’ve replace ‘interact’ in the example, which is one of the ‘partitions’ on odyssey, with ‘resonance’, which I’ve been told is a gpu partition.
Some results
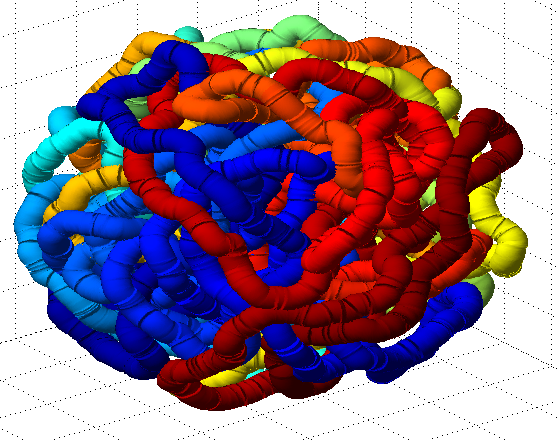
- it seems the unknotted, small box high compression start off with a decent appearance of fractal folding, but even with
GrosbergRepulsiveForce and no truncation, it appears we see crossing later on, both by visual inspection and by running knotAnalysis. Early stages show pronounced color segregation, later stages show more mixing and high (non-integer, troublingly) knot numbers.
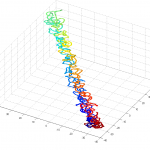
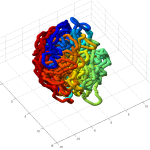

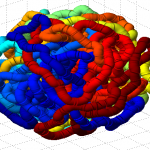
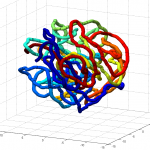
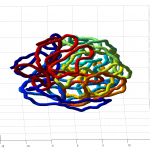
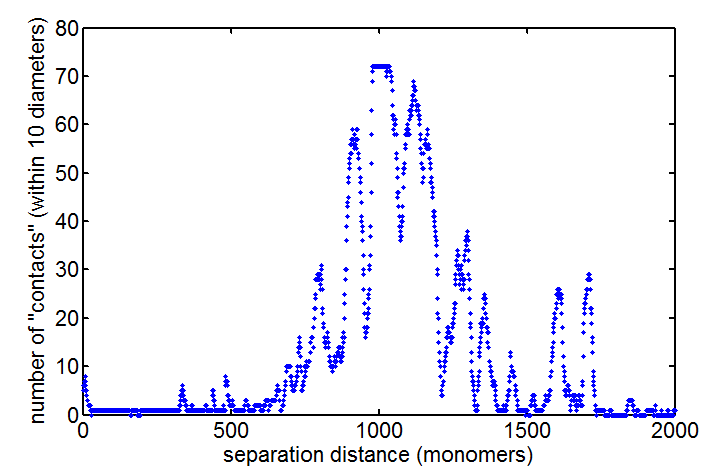
Clearly not simulating for long enough yet to estimate contact frequency (could do more reinitalizations / rounds)