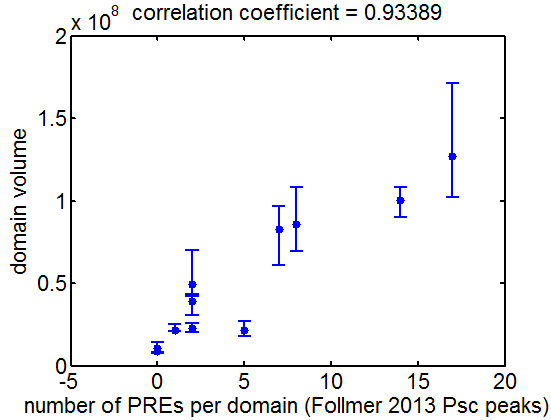
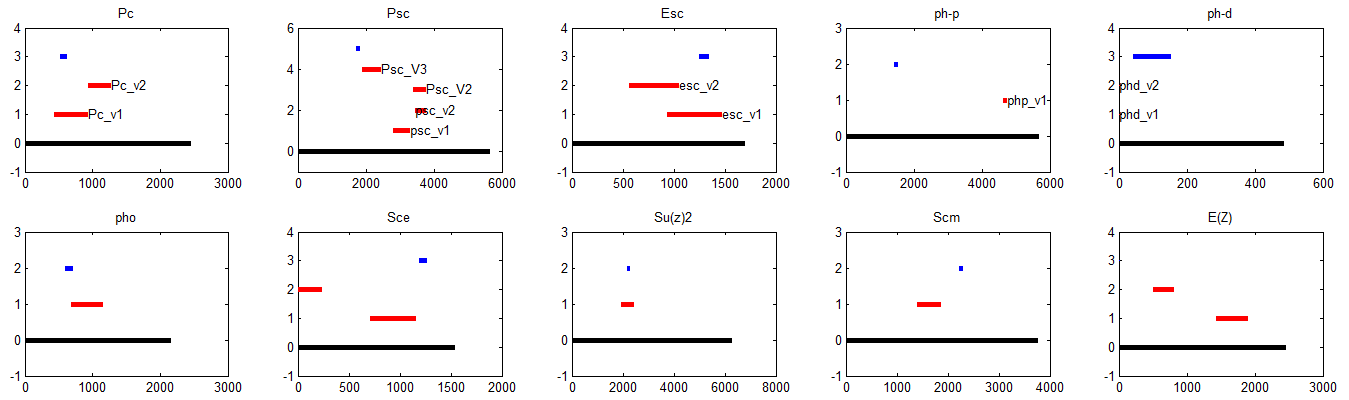
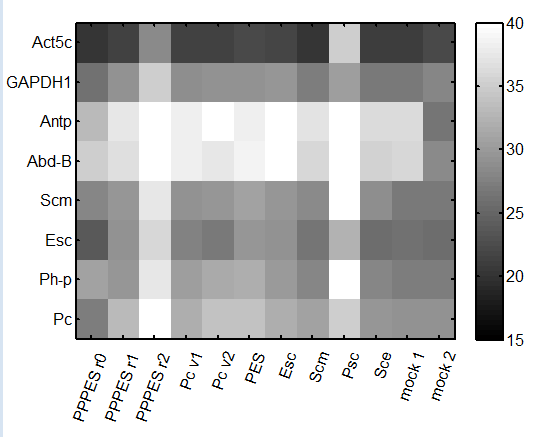
9:30 am – 8:45 pm
Psc deletion
- picked up psc deletion cells from Sonny, along with media
- (also gifted a second aliquot of the Pc-antibody), now frozen.
- also gifted ~200 mL of media ready mixed.
- cells were last plated 7/12 and passaged 1/4. Currently ~60% confluent.
- should remove with trypsin, need to spin down, wash, spin down again, and resuspend to passage.
RNAi experiments
Imaging
- washed cells, 40 min at 60C in 2x SSC
- added 100 nm 540/560 beads at 1:3000 (this density looks great)
- imaging cells – nice and bright, decent density, good contrast. Hopefully these STORM well.
- the TIRF on STORM2 is all kinds of weird
- started new AlistairTemp (2 Tb) for data collection on STORM2 (eSATA is functional)
- installed gitk on storm2 computer
- copy paste of storm2_splitdax imagewriters.py into latest storm2 branch appears to work fine.
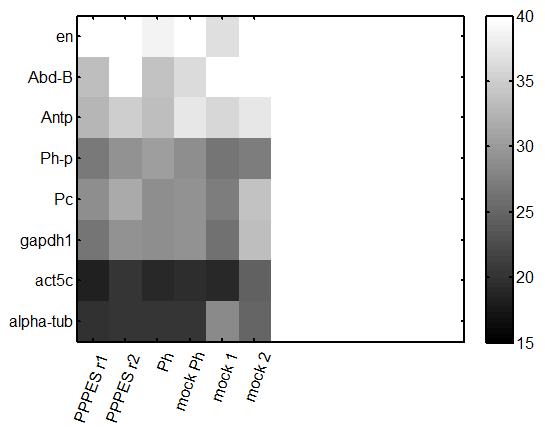
repeat qPCR
- try lower concentration of cDNA – 1 uL per reaction
- primers: Pc, Ph, Abd-B, en, Antp, alpha-tub84, Act5A, gapdh1
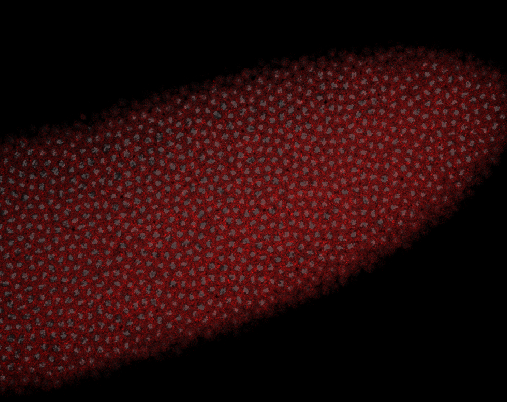
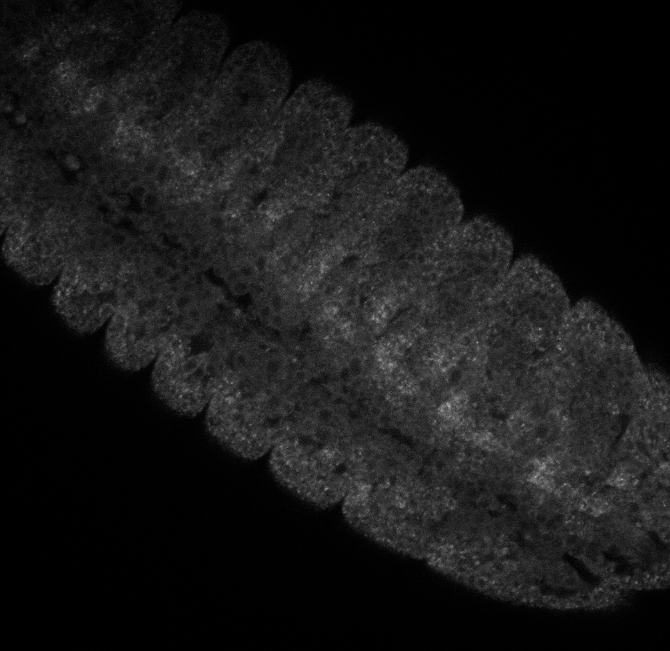
embryo FISH
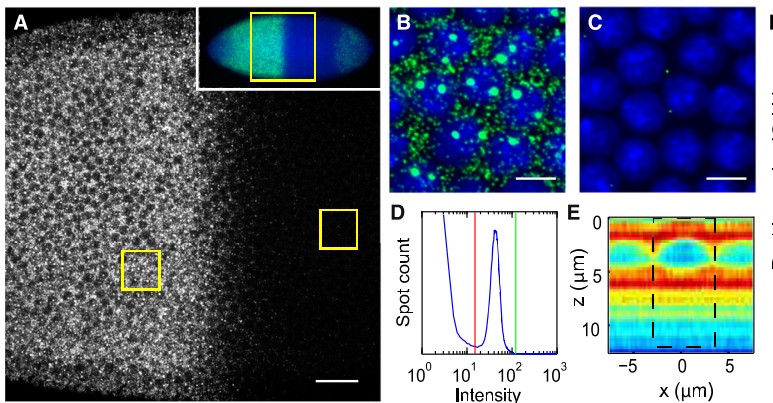
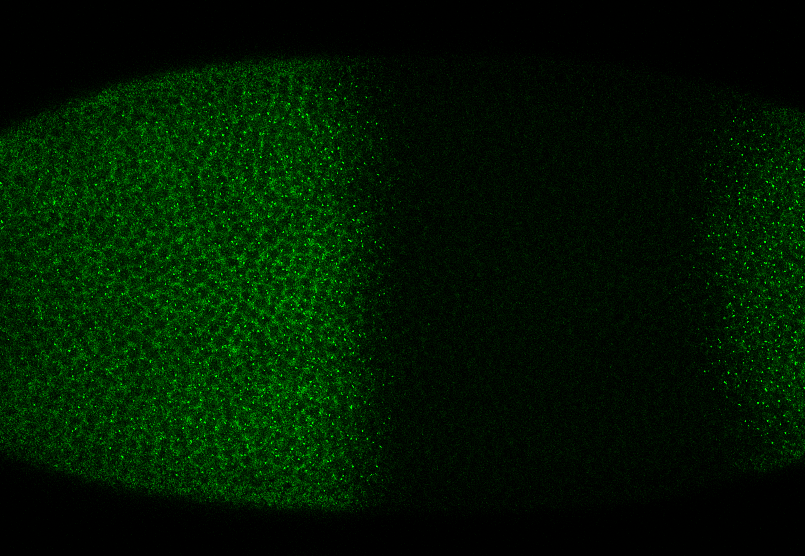
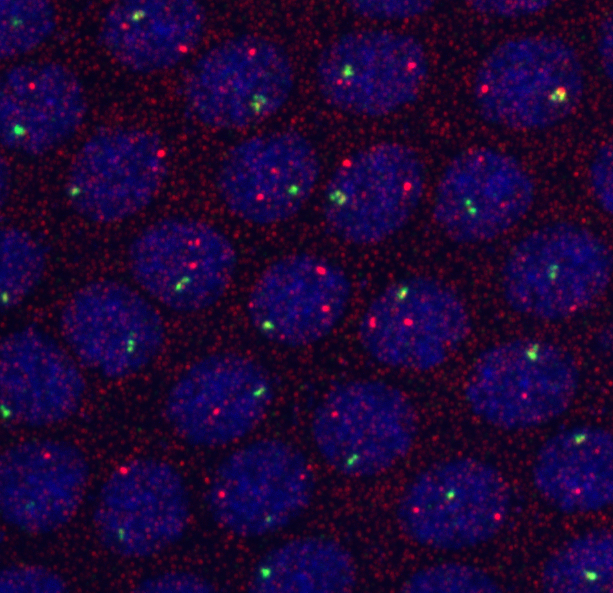
protocol from Little 2013
“Embryos were fixed in5%formaldehyde, 1XPBSfor 20 min, and devitellinated as described by Lecuyer et al. (2008). Fixed embryos were rinsed three times in 1X PBS and washed for 10 min in smFISH wash buffer (4X SSC, 35% formamide, 0.1% Tween 20). Hybridization to probes complementary to the reading frame of hb, Kr, kni,or gt and conjugated to Atto 565 (Sigma-Aldrich; 72464) or Atto 633 (Sigma-Aldrich; 01464) was performed for 16–24 hr at a concentration of about 1nMper probe in hybridization buffer (4X SSC,35%formamide,10% dextran sulfate, 2 mg/ml BSA [NEB; B9001], 0.1 mg/ml salmon sperm DNA [In- vitrogen; 15632-011], and 2 mM ribonucleoside vanadyl complex [NEB; S1402S], 0.1% Tween 20). After two washes of 1 hr in wash buffer, embryos were rinsed twice briefly in 1X PBS, stained with DAPI, and mounted in VECTA- SHIELD (Vector Laboratories; H-1000)”
buffers
- prehybe buffer: 4X SSC, 35% formamide, 0.1% Tween 20 (50 mL master)
- 10 mL 20x SSC
- 17.5 mL formamide
- 500 uL 10% Tween 20
- fill to 50 mL with ddH2O
- hybe dilution buffer: 4X SSC,35%formamide,10% dextran sulfate, 2 mg/ml BSA, 0.1 mg/ml salmon sperm DNA, and 2 mM ribonucleoside vanadyl complex , 0.1% Tween 20 (50 mL master)
- 10 mL 20x SSC
- 17.5 mL formamide
- 10 mL 50% dextran sulfate
- 100 mg BSA
- 500 uL of 10 mg/mL sonicated salmon sperm DNA
- RVC
- 500 uL 10% Tween 20
- would have been better to pre-disolve the BSA in 10 mL of ddH2O — it does not want to go into solution afterwards. heating to 37, will rock O/N.
Chromatin paper
- discuss comments with XZ and BB
- need action items for XZ asap.